
Már Darwin feltételezte, hogy minden ma létező élőlény egyetlen közös ősből származik, ez azóta az élettudományok alapjává vált. Nyilván számos bizonyítékot ismertünk erre eddig is, de most egy viszonylag friss közleményt szeretnék bemutatni, amely a ritkábban emlegetett bizonyítékokkal foglalkozik. Ugye ha egysejtűekről van szó, főleg, ha prokariótákról, akkor nem beszélhetünk kövületekről, mert apró méreténél fogva egy baktériumkövületről semmi sem állapítható meg. Ugyanígy egyedfejlődési sajátosságokról sem beszélhetünk, így marad a ma élő jószágok ismert tulajdonságainak összehasonlítása.
Szerencsére a rendelkezésünkre álló DNS szekvenciák száma folyamatosan gyarapszik, így ma már igazán nagy lefedettségű elemzéseket végezhetünk kizárólag a nyilvános adatbázisokból hozzáférhető adatokkal is. Douglas L. Theobald éppen ezt tette, mégpedig igazán alapos volt. Abból indult ki, hogy attól, hogy két fehérjeszekvencia hasonlósága nem feltétlenül jelenti azt, hogy azok rokonságban is állnak, csak azt, hogy jobban hasonlítanak egymáshoz, mint ahogy azt véletlen szekvenciáktól elvárnánk. Az általánosan használt E érték, amit egy BLAST keresés eredményeként kapunk tulajdonképpen csak annak a null hipotézisnek a valószínűsége, hogy két véletlenszerűen előállított szekvenciát illesztettünk. Hasonló szekvenciák előállhatnak másként is, mint közös leszármazás által, lehetséges, hogy egy adott működést csak egy nagyon szűk szekvenciatér képes elvégezni, így különböző eredetű enzimek szükségképpen hasonlóak lesznek, vagy éppen a mutációs rátát torzítja valami, vagy éppen ha szelekciós nyomás hat egy bizonyos változatra, esetleg a vakvéletlen is létrehozhat két hasonló szekvenciát.
A szerző nem kisebb feladatra vállalkozott, mint hogy olyan statisztikai módszert hoz létre, amivel elkülöníthetőek egymástól a szekvenciahasonlóságok okai, azaz eldönthető, hogy két hasonló szekvencia közös eredetű, vagy valamilyen más okból egyezik. A megoldást Theobald a modellszelekciós elméletben találta meg, a lényeg annyi, hogy több elméletet vesz figyelembe a vizsgálat elejétől kezdve, amelyek között semmilyen különbséget sem tesz a kiinduláskor. Egy modell elbírálásakor alapvetően két tényezőt vesz figyelembe az illeszkedés mértékét és a parszimóniát, amelyek valamilyen szinten egymással ellentétesen hatnak, hiszen az illeszkedés mértéke tetszőlegesen növelhető újabb és újabb tényezők bevonásával, ám a versengő elméletek közül mindig azt választja majd ki, amely kevesebb hasraütéses alapon meghatározott változót tartalmaz. Minden kérdés esetén a három legelterjedtebb modellszelekciós módszert használta a szerző, a „log likelihood ratio” -t (LLR), az „Akaike information criterion” -t (AIC), és a „log Bayes factor” -t (LBF). A modellekben az egyes fehérjeszekvenciák változásai három szabályt követtek: Az egyes aminosavszekvenciák folyamatosan, mutációkkal változtak; Az új gének a régiek duplikációival jelentek meg; A különböző leszármazási ágak változásai nem hatottak egymásra.
A konkrét kérdés, amit föltett pedig viszonylag egyszerű volt: A három nagy élőlénycsoport, az Eukarióták, a Baktériumok és az Archeák vajon egy közös ősből származnak -e? Az elemzéshez huszonhárom általánosan konzervált fehérje szekvenciáit használta, az első elemzésben horizontális génátviteli események figyelembe vétele nélkül tesztelte a szóba jöhető összes változatot, ezek láthatóak az első táblázatban:
ABE= Mindhárom csoport egyetlen közös ősből származik, ez a jelenlegi evolúciós modell
AE+B= Archeák és Eukarióták származnak közös ősből, a Baktériumok függetlenek
AB+E= Archeák és a Baktériumok származnak közös ősből, az Eukarióták függetlenek
BE+A= Baktériumok és az Eukarióták származnak közös ősből, az Archeák függetlenek
A+B+E= Nincs közös ős, mindhárom csoport elszigetelt a többitől
ABE-M+M= A három csoport egy közös ősből származik, kivéve a többsejtűeket, amelyek más eredetűek
ABE-H+H= A három csoport közös ősből származik, kivéve az embert, aki a többiektől független eredetű
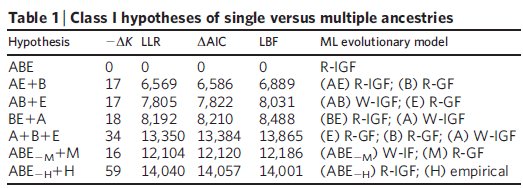
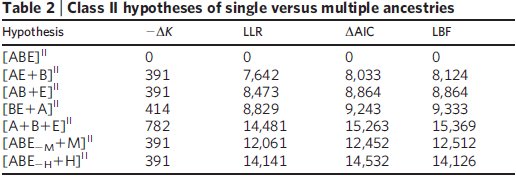
Bár a vizsgált 23 fehérjéből kilencről tudjuk, hogy horizontális génátviteli események is szerepet játszottak a létrejöttükben, meglepő módon az összes csoport egy közös ősből származása így is elég nyilvánvaló az eredményekből, ez a feltételezés nagy különbséggel a legjobb pontszámot kapta a statisztikai elemzés során, mindhárom módszerrel. A közös ősből származás e mellett az adatokra legjobban illeszkedő modell és a legegyszerűbb is, úgyhogy mindkét, látszólag egymásnak ellentmondó szempont szerint egyszerre a legjobb magyarázat az adatokra. Ha a modellbe bevezették a horizontális génátvitelt is, ahol az egyes gének szabadon átkerülhettek bármelyik ágról bármelyik másikra, az egyes ágak összeolvadhattak, jóval bonyolultabb leszármazási fákat hozva létre ezzel, nem túl meglepő módon sokkal nagyobb különbséggel győzött a közös őst feltételező modell, vagyis a statisztikai elemzés alapján egyértelműen ez illeszkedik legjobban az ismert adatokhoz és nem mellesleg ez a legegyszerűbb modell is, ez látható a második táblázatból.
Miért érdekes ez a közlemény? Egyrészt egy tisztán statisztikai, előfeltételezésmentes módszerrel megerősíti, hogy a földi élőlények egy közös ősből származnak. Ez nem igazán újdonság, eddig is tudtuk. A módszer előnye, hogy ugyanígy vizsgálható lenne bármilyen más modell is, például az evolúciótagadók ugyanebbe a vizsgálatba beilleszthetnék a saját modelljeiket (ha sikerülne végre alkotni egyet) és ugyanezzel a statisztikai módszerrel azonnal vizsgálhatóvá válna a valószínűsége is.
Theobald D.L.(2010): A formal test of the theory of universal common ancestry; Nature 13;465(7295):219-22.
Sexcomb